R函数 | 写个自己的write_fasta()导出fasta序列
介绍
昨天推文写到通过refdb::refdb_import_NCBI() 下载了序列,是一个数据框格式。如果我想把序列写出为fasta格式,那么该如何做呢?
这里自己写个write_fasta() ,但是用类似write_fasta() 这个函数名在各大包中占用的太多了,我把名字改为了write_bio_fasta() 。
环境准备
其实下面都是通过包名::函数 方法调用的,写出来主要是让大家看下用了什么包。
1
2
3
4
5
6
7
|
rm(list = ls())
library(refdb)
library(dplyr)
library(stringr)
library(tidyr)
library(readr)
library(magrittr)
|
准备数据
这里还是用昨天推文提到的refdb::refdb_import_NCBI()
1
2
3
4
5
6
7
|
# 1. 下载序列
silo_ncbi <- refdb::refdb_import_NCBI(query = "Silo COI",
full = TRUE,
max_seq_length = 10000,
seq_bin = 200,
verbose = TRUE
)
|
处理序列数据
上面的silo_ncbi 对象是个数据框,我们从中提取id,sequence,species,length四列,作为要输出的fasta信息。前两列分别为必需的fasta id和fasta 序列,这里我们合并后两列,作为 fasta的描述部分。描述列为非必须部分。
注意:列的顺序不要改;列名可以随意。
1
2
3
4
5
6
|
# 2. 准备序列数据框
# 准备要写出的序列,数据框格式,第一列为id,第二列为sequence,第三列为description
## 这里将species和length两列内容合并作为description部分。
simple_seq <- silo_ncbi %>% dplyr::select(id,sequence,species,length) %>%
tidyr::unite(col = "description",species,length,sep = " ",remove = TRUE, na.rm = FALSE)
# unite 合并数据框的列
|
simple_seq 内容如下。
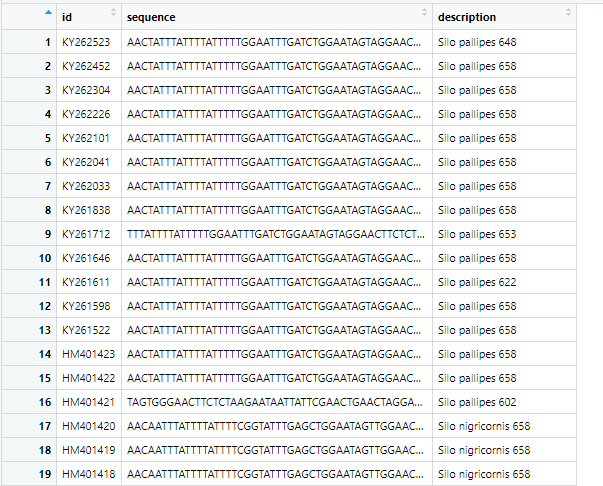
其实,也可以完全把第一列id和第三列description 合并到第一列。
参数说明
data 一个数据框,含2列或者3列。具体格式详见处理序列数据 部分。 file 需要输出的 fasta文件。width = 60 输出结果fasta序列每行碱基数append = FALSE 和num_threads = readr_threads() 同write_lines() 的参数
函数定义
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
|
# 3. 定义fasta序列写出函数:write_bio_fasta()
# simple_seq 三列的列名,不重要。
write_bio_fasta <- function(data, file, width = 60, append = FALSE,num_threads = readr_threads()){
# check data
stopifnot(is.data.frame(data)) #必须为数据框格式。
n_col <- ncol(data)
if(n_col != 2 & n_col != 3){
stop("`data`: the input must be a data frame with 2 or 3 columns!")
}
# prepare the list for `write_lines()`
fa <- list() #提前定义下
#width = 60 # 输出序列的的width
for (i in seq(nrow(data))){
n = length(fa) + 1 #向fa list 添加到第n个元素,也就是fasta序列文件的行数了。
if(n_col == 2){
id <- data[i,1] %>% stringr::str_replace(pattern = "^", ">") %>% as.character()
fa[[n]] <- id
} else { #把esle if判断n_col = 3去掉了,也没写不等于2或3时如何。因为前面已经限定过了。
id <- data[i,1] %>% stringr::str_replace(pattern = "^", ">") %>% as.character()
description <- data[i,3] %>% as.character()
fa[[n]] <- paste(id,description,sep = " ")
}
seq <- data[i,2] %>% as.character() #refdb的结果中seq列并不是简单的向量,转一下。
for (x in seq(ceiling(stringr::str_length(seq)/width))) {
fa[[n+1]] <- stringr::str_sub(seq,start = ((x-1)*width+1), end = x*width)
n=length(fa)
}
}
# 写出序列,设置append与num_threads
readr::write_lines(fa,file = file, sep = "\n", append = FALSE, num_threads = readr_threads()) #要求为list或向量。若为list,写出的list内容,不含name
}
|
执行
1
|
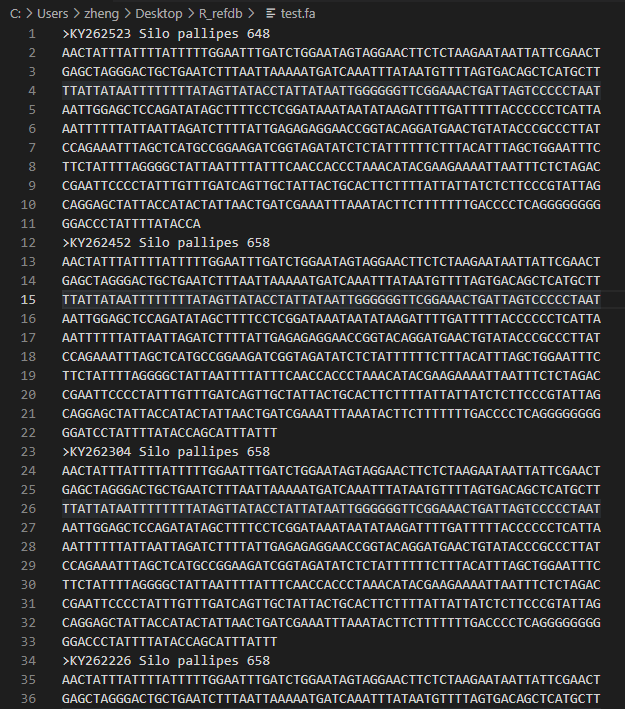
write_bio_fasta(data = simple_seq, file = "test.fa",width = 70)
|
序列内容如下:
小结
有write_bio_fasta() 就该有read_bio_fasta() ,但是临近毕业,我在尽量克制自己做“与毕业无关的事”。暂时不写这个函数。其实write_bio_fasta() 也是个与毕业无关的函数,只是没控制好好奇心,还是写了下。